BBI Contributing Writer
Combination products those products that contain combinations of drugs, devices or biologics have endured a convoluted regulatory path to approval for many years. New developments in pharmacologics, biotechnology, genomics and proteomics have yielded products to deliver drugs or biologics to specific targets within the body, resulting in unique scientific, policy and regulatory issues. The official designation of a combination product includes two or more regulated components that are physically, chemically or otherwise combined, packaged together or provided separately where both are required to achieve the intended use.
Previously in the U.S., when a company filed for regulatory approval of a combination device, separate submissions were sent to the FDA centers that governed each of the individual components the Center for Drug Evaluation and Research (CDER), the Center for Devices and Radiological Health (CDRH) and the Center for Biologics Evaluation and Research (CBER). Each center has different time requirements and procedures for conducting reviews; thus, the centers worked somewhat independently and lacked the strategic coordination necessary to avoid delaying the product approval. In addition, concerns about the need for consistency and predictability in the assignment to a particular agency and the absence of clarity in post-market combination products regulatory controls created apprehension. Whether this confusion was driven by product innovation, FDA staff turnover, lack of communication between the various centers and a reviewer simply not knowing where to send an application for consult with another agency, or a myriad of other reasons is not clear. It is agreed that until recent months, the FDA regulatory review pathways that were developed decades ago, and revised in 1991, did not include appropriate provisions for the exploding number of combination products being presented for review.
Company management teams developing a combination technology have undertaken increased responsibility to determine the most expeditious path to regulatory approval and marketability. Careful review of the regulations, some strategic regulatory management, discussions with the proposed FDA center's product jurisdiction officers, leverage of any preceding combination products that have already received regulatory approval and in some cases just plain luck have allowed some combination products to be approved. One product, the Cypher drug-eluting coronary stent from the Cordis (Miami Lakes, Florida) unit of Johnson & Johnson (New Brunswick, New Jersey), approved with much fanfare in April, is a combination device that was assigned to CDRH (device) and had a cadre of regulatory recruits working in addition to top-down management emphasis to secure approval. The clinical success of this product increased awareness of combination product regulatory approval at a time when significant changes were concurrently in motion at the FDA. For companies without the well-staffed infrastructure to marshal their product through the FDA labyrinth, the changes at the agency are especially welcome.
Combination products also include such products as orthopedic implants with growth factors, biologically based sealants, glues and hemostatic agents, pain management pumps, interferon injector pens, photodynamic therapy, antimicrobial coated catheters, living cells encapsulated in a semipermeable membrane that secrete various biological substances and many more. Additionally, with drugs that correspond to $37 billion in revenue each year coming off patent soon, according to the North Carolina Biotechnology Center (Research Triangle Park, North Carolina), drug delivery and combination products will be an important wave of products in the near future. At a Combination Products conference, held in July in Washington, developed and managed by Barnett Educational Services (Media, Pennsylvania), a division of Parexel International (Waltham, Massachusetts), methods to shorten the time to product approval in the U.S. and internationally were revealed by industry professionals from around the globe.
Office of Combination Products
The new FDA Office of Combination Products (OCP) was initiated Jan. 1 and the final ruling guidance documents were published in late June. The OCP is part of the Office of the FDA Commissioner and functions as an "umbrella" over all the centers pertaining to combination products. The OCP determines which center is responsible for lead review of a combination product on a product-by-product, case-by-case basis. Mark Kramer, director of the OCP, has commented on the role of the new office, saying, "The OCP is responsible for assigning the appropriate FDA Center for review of a specific combination product based on the primary mechanism of action. The OCP will not perform the actual review of these complex products as that will be left for each respective center once the product is assigned."
The key responsibilities of the new office are 1) the prompt assignment of combination products to agency centers, 2) timely and effective premarket review of such products, 3) consistent and appropriate postmarket regulation of similar products and 4) annual reports submitted to appropriate committees of Congress on the activities and the impact of the office. Also, with the initiation of the OCP, combination products now are included in the FDA tracking database, which allows company personnel to monitor their regulatory progress toward approval in a systematic manner. This was previously possible only with products reviewed under a single FDA center.
Jonathan Kahan, partner in Hogan & Hartson, a Washington law firm, commented, "The good news from the new OCP initiative is that the FDA has a heightened sensitivity to combination products. There is a new and welcome recognition that innovation in combination products should not be stifled by regulatory inefficiencies. The FDA is really interested in ways to make the process work more efficiently." For investors in development-stage companies who want to project the value of a medical device or a drug company slated for possible investment, this new infrastructure may better predict when a U.S. revenue stream is probable.
Table 4 below outlines the agency within the FDA designated to review the product based on the primary mechanism of action. Most of these products have not enjoyed the projected efficiency of the OCP, and thus have had lengthy approval processes.

Privately held EKOS (Bothell, Washington) received 510(k) clearance in July for its micro-infusion ultrasound catheter designed to diffuse therapeutic agents into the vasculature. Based on the mechanism of action, ultrasonic dispersion of the infused fluid, this catheter was approved not as a combination product, but just as a device. Jocelyn Kersten, EKOS regulatory manager, said that "Thorough research and careful testing confirmed EKOS's position that therapeutic agents infused through our catheters were neither positively nor adversely affected by the ultrasound energy to which they are exposed. The resulting 510(k) clearance confirmed our strategic approach to obtaining marketing clearance." Understanding the regulations regarding combination products allowed the manufacturer to position its device as a stand-alone product and design definitive testing to support their position.
The FDA review requirements are significantly different as a stand-alone device vs. a combination device or drug/biologic. Even the assignment of a combination product to CDRH rather than to CDER or CBER will significantly shorten the time to market for the product and decrease the costs. Table 5 (below) highlights some of the differences between the device and drug or biologic approval process.
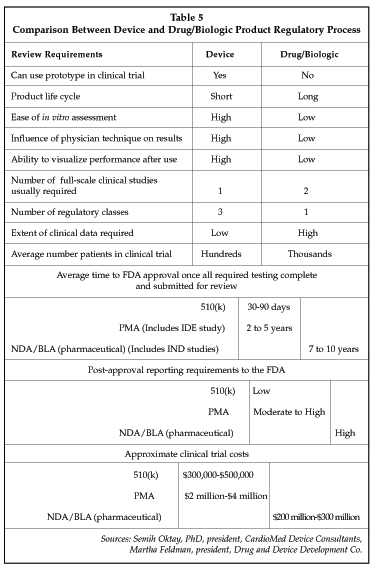
Combination product trials
Clinical trials for combination products will inherently be more comprehensive, costly and complicated than those required for individual drugs, devices or biologics. According to Barbara Fant, PharmD, president of Clinical Research Consultants (Cincinnati, Ohio), "The clinical trials to evaluate the safety and effectiveness of a combination product must take into account the individual properties of each component, as well as the properties of the components when used in combination." For example, she said, "pharmacokinetic studies for the recently approved drug-eluting stent had to be performed on the drug alone as well as the combination product in patients who had one or more drug eluting-stents implanted." Additionally, Fant noted, "drug interaction studies for the drug alone, one first-in-man study, one pilot study and one large randomized pivotal study were conducted to support the stent's approval." She added that the FDA "also required a post-marketing study to evaluate long-term safety and effectiveness outcomes."
There are important differences between clinical trials for drugs/biologics and devices in adverse event reporting, mechanisms of action and regulatory requirements. Clinical and statistical experts with experience in both components of the combination product can maximize success.
Determining FDA center of authority
If previous FDA decisions for similar products are not available, and a definitive path to a specific agency is not clear, the applying company can file a Request for Designation (RFD) with the Office of Combination Products and select which center it proposes for primary jurisdiction. Even if the decisions and paths are defined, but are not consistent with the company's position or goals, an RFD can be filed to try to differentiate the product. In the RFD, the company can outline its positions and strongest justifications regarding the regulation of the product. The RFD includes an overview of the product, any completed testing, proposed indications for use, dose and route of administration for the drug or biologic, and other pertinent information. The outcome of the RFD and follow-on negotiations will determine the lead center for review, consulting center if appropriate, clinical data requirements, submission requirements and even post marketing regulation.The company should file an RFD before filing any application for premarket review. If the Product Jurisdiction Officer has not informed the company within 60 days of filing, the company's positions and recommendations become the designated regulatory pathway.
Lee Leichter, president of P/L Biomedical Consulting (Fort Myers, Florida), who served as chairman and moderator of the Combination Products conference, said, "It is important to evaluate the regulatory and jurisdictional issues at the product development stage to develop product strategy early. If a company waits until the initiation of clinical studies to determine how the FDA will regulate a combination product, significant time and costs can be added to the approval process." When meeting with the FDA, it also is important to keep in mind that the reviewing division may be aware of issues related to similar products. Although the challenges and strategy of a particular competing product are not public information, the specific reviewing FDA agency can provide guidance based on problems competitive products have experienced.
The Cypher stent was approved through CDRH, which was designated as lead reviewing center with CDER consultation. Table 6 below focuses on the risk of approval based on the required pre-clinical and clinical studies to support the approval of such products.
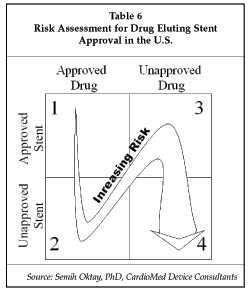
The level of risk increases as the numbers increases from Block 1 to 4. Companies with a product in Block 1 are able to leverage information (pre-clinical and clinical) from previous experience/applications. Companies with a product in Block 4 will have high requirements for clinical trials and product testing to demonstrate safety and effectiveness. Semih Oktay, PhD, president of CardioMed Device Consultants (Gambrills, Maryland), a former reviewer in the Interventional Cardiology Devices Branch of CDRH, provided further comment on the required testing. "Typically, for a drug-eluting stent application, the company should provide chemistry, manufacturing and controls (CMC), systemic preclinical pharmacology and toxicology studies, systemic clinical, stability and shelf-life data to demonstrate safety of the combination product. Furthermore, to demonstrate reasonable assurance of safety and effectiveness, animal and clinical study requirements will also change based on where a company's product is within this 2x2 matrix." Additionally, the FDA recommends that companies make use of existing guidance documents from both CDRH and CDER to fulfill the preclinical safety requirements.
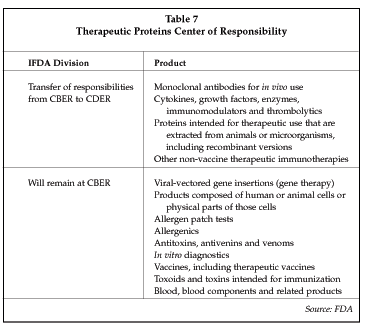
Also included in recent FDA changes is the transfer of therapeutic products to the CDER (drugs). The regulatory responsibility, review and continuing oversight for many biologic therapeutic products are being transferred from CBER (biologics) to the CDER. This change in regulatory responsibility will result in the transfer of applications of many product classes as shown in Table 7, such as medical devices with recombinant proteins.
User fees for medical devices
FDA user fees paid for FDA reviews of all medical devices, including combination products, were initiated in 2003, and increases for 2004 were announced July 31 and are shown in Table 8 below. The increased amount takes effect with the 2004 fiscal year for submissions received on or after Oct. 1. The fees are used to support the FDA's long-term goal of making substantial improvements in device review performance. According to Frank Claunts, FDA user fee finance coordinator, increases are due to several factors including an increase in target revenue to accelerate the review process, an adjustment for inflation and an adjustment for an unforeseen revenue deficit in FY03 due to an approximate 30% shortfall in anticipated premarket approval submissions, both initial and supplements.
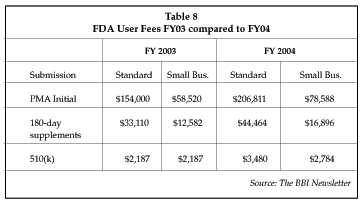
The first time a small business, defined as revenues equal to or less than $30 million, submits a PMA, it is exempt from paying user fees, and it maintains reduced fees while below that revenue level. Some companies with eligible 510(k) products may select third-party 510(k) review in lieu of FDA review. Third-party submissions are exempt from the FDA user fees; however, third-party review companies charge similar or higher fees for regulatory review. In comparison, FDA user fees for human drugs and biological products have been required since 1992 and for 2004 are $573,500 for an application requiring clinical data, and $286,750 for an application not requiring clinical data or a supplement requiring clinical data, $226,800 for each manufacturing establishment, and $36,080 for each product sold.
European regulatory path
Within Europe, there is no regulatory agency dedicated to defining specific regulatory paths for combination products. European approval requirements place greater emphasis on safety and performance; and although clinical efficacy requirements are included, they are not necessarily as robust as those in the U.S. Devices in general are classified based on a number of rules described in the Medical Devices Directive, which builds on the concept of a risk-based approach related to the device's duration of use, invasiveness, and associated hazards. A product can only be regulated as a medicinal (whether drug or biologic) or a device. Once approved for market, a product can be sold anywhere in the European Economic Area (the original 15 EU countries and after May 2004, the additional 10 new member states, plus Iceland, Liechtenstein and Norway). Other countries throughout the world also accept the CE mark.
A medicinal product cannot be CE-marked. It can only be marketed through submission of a Marketing Authorization Application (MAA) either to the governmental authority in a single country, referred to as the Competent Authority, or to the European Medicines Evaluation Agency (EMEA; London), a centralized agency that can authorize a medicinal product throughout the EU. The equivalent of a Competent Authority in the U.S. is the FDA.
These two different paths one of which results in a CE mark (medical device) and the other with a Marketing Authorization (medicinal product) have resulted in the same product from different manufacturers who chose different routes to approval, being CE-marked in one instance as a medical device and in another instance approved as a medicinal product. Additional confusions arise when a product from a single manufacturer is CE-marked as a medical device but is still considered a medicinal product in some countries.
An example of such a product is BrightStent from Angiogene (Montreal, Quebec), a drug-eluting stent with an unapproved radiopharmaceutical drug. According to Guy Chamberland, PhD, Angiogene vice president for regulatory affairs and drug development, "The addition of an unapproved drug to a device brings on new regulatory challenges in Europe. The company must now ensure that the product submission technical file also contains information that will meet the requirements of the medicinal competent authority such as the EMEA."
Since similar advantages of reduced cost and time to market apply for devices over medicinals in Europe, the ability to select the least-burdensome route to approval is advantageous. Also, for some medical device manufacturers, a determination that a product is to be regulated as a medicinal product (drug or biologic) may mean that product is outside the company's legal, financial or product distribution capabilities.
Selection of Notified Body critical
In order to market any medical device in the EU (except lowest-risk Class 1 devices such as non-invasive, non-active products like crutches), the manufacturer must work with a Notified Body. Notified Bodies are independent companies designated by the Competent Authority in each EU country that have responsibility for the regulation of medical products. Each Competent Authority can designate one or more independent Notified Bodies within their country to act as agents to review, audit and certify (CE mark) medical devices.
The European Commission currently lists 68 authorized Notified Bodies; however, only a small segment of these have experience with combination products. Selection of one of these Notified Bodies can significantly expedite the path to approval for a combination product. It also is important to ensure that the Notified Body a company selects concurs with that company's determination about how the product will be regulated.
To ensure the most expedient approval, the Notified Body selected should have scientific and technical expertise and specific experience with a product as closely aligned with that company's product as possible and adequate resources to perform required functions. The Notified Body should have a good relationship with the pharmaceutical Competent Authority required for certification consult.
According to Leichter, "If the pharmaceutical substance combined with your device is a newer, proprietary product and you select the Competent Authority that was used for the drug approval, that agency should be experienced with and have easy access to the Drug Master File." Other considerations are whether the Competent Authority has had any experience in these types of consultations and has the organization, formal procedures, expertise and resources to prevent delays. Many Competent Authorities are not organized or staffed to move this process forward on timelines acceptable to medical device companies.
One distinction important to remember is that it is not necessary for a company to use the same Notified Body for all of its products. If a company is adding a different type of product to its portfolio, it is important to reevaluate the Notified Body selection for that product.
Once a company selects a specific Notified Body for a particular product, it becomes difficult to change to an alternate Notified Body for that product thus the importance of thoroughly investigating before making a formal application for a particular Notified Body. One can investigate their capabilities, obtain a quote, and even request a meeting in person; however once a formal application is made, it is difficult to change should the company become dissatisfied. Thus, management should view the selection of a Notified Body just as it would any important contractor and assure that all of the experience and qualifications that will expedite the process.
It is obvious that the need for medical devices to deliver drugs and therapeutic biologics will result in many more combined products. The Office of Combination Products and evolving path within Europe offer a company with an emerging combination product the unique opportunity to help determine the most likely and acceptable path to approval.
The best strategy for regulatory approval in both the U.S. and Europe is for the company to initiate regulatory planning very early during the design process and assemble the right team with expertise in both drug and device development. By researching the current directives, guidance documents and any available precedents the company may be able to influence the way the product is regulated. At a minimum, any company that uses these resources to establish and support a regulatory approval route will increase its chances to negotiate one that is consistent with its own business objectives.