The interventional sector of cardiology faces new challenges and questions concerning DES — and stenting in general — as companies seek to bolster percutaneous therapies and push new devices and technologies into this arena
Cardiovascular Device Update, Contributing Editor
NEW ORLEANS — The cardiology device market — already feeling the pressure of increasing concerns about the safety of drug-eluting stents (DES) and the failure of implantable cardioverter defibrillator (ICD) technologies — suffered another blow here at last month’s annual conference of the American College of Cardiology (ACC; Washington, DC).
Over the past six months the DES market has retreated from the highs it reached last year, stabilizing at a level of about $6 billion worldwide. Now, new data issued at the sessions from the COURAGE trial threatens to at least temper the odds of growth resuming in the near future — both for DES and the whole coronary stenting arena — and has raised the specter of further declines if cardiologists decide to change their practice patterns based on the results of that trial.
Against the backdrop of this considerable pushback, the sessions were successful at doing what conferences do: offering a venue for unveiling of new products, some of these promising to resolve the safety issues with DES and eliminate other drawbacks of the technology. Cardiologists are also continuing to expand their use of device-based therapies to encompass additional applications in stroke prevention, treatment of peripheral vascular disease, treatment of heart failure, and repair and replacement of heart valves. The growing diversity of this market will provide added stability in the future. For suppliers, the market will continue to be challenging, as the pace of innovation is not abating in spite of the increasing challenges of new product development.
DES faces regional declines
As shown in Table 1, the most recent data concerning DES utilization shows a somewhat mixed picture, depending on the region of the market analyzed. Utilization dropped dramatically in the U.S. during the latter part of 2006, following the widely publicized results of a metanalysis presented by Camenzind et al. at the 2006 World Congress of Cardiology conference in Barcelona in September and highlighting issues with late stent thrombosis, and the subsequent notice from the FDA regarding the increased possibility of late thrombosis in off-label uses of DES.
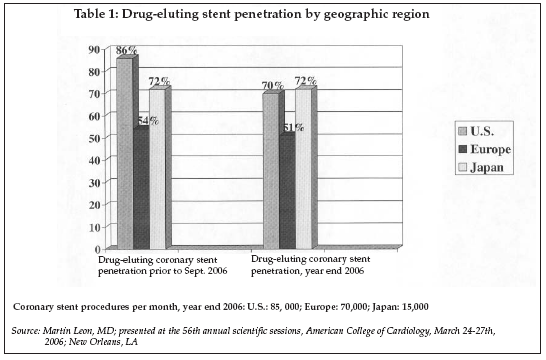 |
In the U.S., which has the highest volume of stent procedures, the penetration of DES, which had reached about 86% at its peak, has now dropped to 70%. In Europe, however, there has been only a slight decline in penetration, while in Japan, utilization has climbed steadily since DES technology was introduced, and is now at 70% of coronary stent procedures — the same as in the U.S. with no impact of the announcements regarding late stent thrombosis.
One apparent reason for the minor impact of reports of stent thrombosis on utilization in Europe may be that extended dual anti-platelet therapy (for nine months) has been the norm there for most patients receiving DES devices. In contrast, the U.S. the extension from the original recommended interval of three months out to one year was made only recently. While the exact relationship between the duration of dual anti-platelet therapy and the risk of late stent thrombosis is not yet well-defined, extending the therapy interval should reduce the risk of thrombosis.
Other factors believed to be related to stent thrombosis risk include the number of stents placed, total stent length, the occurrence of renal failure and other complications and co-morbidities, and lack of complete apposition of the stent struts to the vessel wall.
Late thrombosis is still not a well-understood phenomenon, however, and assessment of the true degree of risk posed by late thrombosis is made more difficult by equivocal results of studies conducted to date to assess the issue.
Mortality risk widely publicized
For example, widely publicized data from the Swedish Coronary Angiography and Angioplasty Registry, published in early March by Bo Lagerqvist, MD, PhD, et al. in the New England Journal of Medicine, showed an increased risk of death after six months for DES compared to bare metal stents (BMS).
The recommended duration of dual anti-platelet therapy for patients in Sweden during the period of the study was six months. However, results from another registry in neighboring Denmark, presented at the ACC conference in a late-breaking clinical trial session by Michael Maeng, MD, of Aarhus University Hospital (Skejby, Denmark), showed that, for patients treated with dual anti-platelet therapy for 12 months, there was no difference in stent thrombosis up to 12 months for patients receiving drug-eluting stents compared to those receiving bare metal stents.
While there was a small but statistically significant increase in thrombosis and myocardial infarction 12-15 months after discontinued dual anti-platelet therapy, the researchers concluded that the minor increase in risk of late events did not outweigh the benefits of DES in reducing restenosis. Target vessel revascularization was reduced by 42% in the DES group compared to the BMS group.
There is no doubt, however, that safety concerns have had a negative impact on DES use, particularly in the U.S. As discussed by John Hodgson, MD, of St. Joseph’s Hospital and Medical Center (Phoenix), about 40 stent thrombosis events are now reported annually to the FDA, a low number compared to the 1 million patients per year who receive stent implants — nevertheless a cause for concern.
Data cited by Hodgson published in 2006 shows that the rate of late stent thrombosis more than doubles, from 1.7% to 4.4%, if dual anti-platelet therapy is stopped, creating a management challenge for physicians who must work with their patients to try to insure that they maintain drug regimen compliance.
Questions attract next-generation solutions
Safety issues with existing DES have fueled development of a wide range of next-generation devices aimed at resolving the problems of late stent thrombosis and hypersensitivity reactions to stent polymers while still preserving the advantages of low restenosis rates.
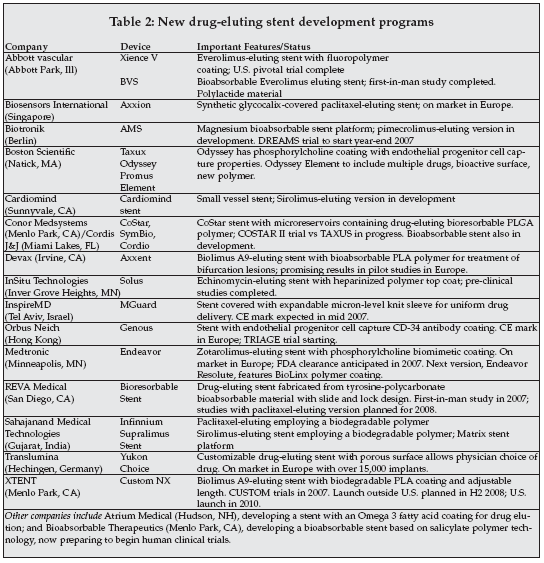
As shown in Table 2 (following page), existing suppliers of DES, as well as emerging companies in the cardiovascular device arena are developing next-generation DES devices employing technologies such as bioabsorbable polymers, multiple drug elution, bioabsorbable stents, advanced stent designs, bioactive surfaces, and new drugs. Bioabsorbable polymers and stents are a major focus of development, conceptually eliminating issues with long-term implants and their effects on vascular tissue.
 |
The Abbott Vascular (Abbott Park, Illinois) unit of Abbott Laboratories (Abbott Park) is one of the leaders in development of bioabsorbable stents, based on initial results from the ABSORB trial, which evaluated the Bioabsorbable Everolimus-Eluting Coronary Stent System (BVS EECSS) originally developed by Bioabsorbable Vascular Solutions (Mountain View, California), a former subsidiary of Guidant, now a unit of Abbott.
As discussed by Patrick Serruys, MD, PhD, of Erasmus Medical Center (Rotterdam, the Netherlands), only about three to six months of mechanical support of a stented vessel is in principle needed in order to prevent restenosis, allowing use of a bioabsorbable device to eliminate polymer effects, fracture issues, and problems with MR and CT imaging.
The device evaluated in the ABSORB trial consists of a polylactide backbone and a bioabsorbable polylactide coating containing everolimus. The release characteristics of the drug are similar to those for the Cordis Cypher stent. At six month follow-up, late loss in a group of 30 treated patients was 0.44 mm, a figure midway between the loss for Abbott’s Xience V drug-eluting stent and a BMS.
There were no target lesion revascularization events in the 30 patients, demonstrating that while late loss was relatively high, it was not sufficient to affect clinical outcome. Importantly, there were no stent thrombosis events, although the follow-up interval is of course too short to allow late stent thrombosis to be evaluated.
According to Serruys, data from intravascular ultrasound follow-up studies shows that the BVS stent is being absorbed. In addition, Abbott has developed a modified mechanical design expected to reduce late loss by 50%, to a level of 0.2 to 0.25 mm, approaching that for the Xience V stent. Although long-term data will be needed to determine if use of bioabsorbable materials eliminates late stent thrombosis, the data so far are promising, particularly if the modified design lowers late loss closer to the level now obtained with the latest-generation drug-eluting stents.
Other companies developing bioabsorbable stents include Cordis (Miami Lakes, Florida), Biotronik (Portland, Oregon), REVA Medical (San Diego), and Bioabsorbable Therapeutics (Menlo Park, California).
While bioabsorbable stents appear to address many of the drawbacks of the current generation of DES, there are nevertheless a number of issues that must be addressed, as discussed by Ron Waksman, MD, of Washington Hospital Center (Washington, DC), including the design of scaffolding with adequate radial strength, control of the rate of degradation, biocompatibility of the polymer and its breakdown products, residual material remaining in the tissue, control of drug elution if a drug is used, and radiopacity.
Various hybrids being readied
A number of companies are developing stents with bioabsorbable polymer coatings, a hybrid approach to bioabsorbable device development that retains the long-term mechanical support provided by a BMS framework but eliminates long-term exposure to polymeric materials, in principle avoiding one of the primary causes of late stent thrombosis.
Examples include Conor MedSystems (Menlo Park, California) CoStar and other versions of that technology, being acquired by Cordis; the Devax Axxent stent, a bioresorbable stent from REVA Medical; stents with bioresorbable polymers from Sahajanand Medical Technologies (Surat, India); the bioabsorbable oil coating being developed by Atrium Medical (Menlo Park, California); and the Custom NX device from Xtent (Menlo Park, California)
Initial data from studies with the CoStar stent, discussed at the ACC meeting by Ron Waksman, MD, of Washington Hospital Center, are promising, with no stent thrombosis observed at six months and at 12 to 24 months, but the number of patients is small so there is as yet not definitive proof that combining bioabsorbable polymers with a metal stent framework solves the issue of late stent thrombosis.
Yet another approach being developed by multiple companies is bioactive surface technology for stents. Examples include the glycocalix coating from Biosensors (Singapore), the endothelial progenitor cell capture coating developed by OrbusNeich (Hong Kong), and new devices in the Odyssey family being developed by Boston Scientific. In principle, such devices may allow the use of anti-proliferative drugs to be avoided, since the DES coating is designed to promote growth of healthy endothelium, avoiding neointimal cell proliferation that leads to restenosis.
Results for patients treated with the OrbusNeich Genous stent in the 3,000+-patient eHEALING registry, however, indicate that rates of stent thrombosis, including late stent thrombosis, are similar to those observed with existing drug-eluting stents such as the Cypher.
As discussed by Robert de Winter, MD, of Academic Medical Center (Amsterdam), a 0.8% stent thrombosis rate is observed at eight months with the Genous stent, and rates of major adverse cardiovascular events are 6.7% for the Genous stent, compared to about 4.6% for Cypher at the equivalent time point.
Various factors impacting bioactivity
One factor affecting the performance of bioactive stents is the responsiveness of the patient’s cells to the active coating. Studies with the Genous stent have shown that the rate and degree of endothelial covering depends on the level of circulating endothelial cells, which in turn can be affected by factors such as whether the patient is taking statin drugs. In addition, the functionality of circulating endothelial cells is a variable that can affect performance.
In spite of the challenges facing the DES market, suppliers are forging ahead with development of next-generation DES devices.
Data on the SPIRIT III pivotal trial of the Abbott XIENCE V stent was presented at the conference by Gregg Stone, MD, of Columbia University Medical Center (New York), with some analysts projecting that the Xience will gobble up the major market share in the DES sector, assuming approval, with roll-out likely in 2008.
XIENCE V is a next-generation device consisting of a cobalt-chromium alloy stent framework covered by a fluoropolymer layer, with Everolimus the eluted drug. XIENCE V produced rates of target vessel revascularization that were 60% lower than for the TAXUS stent, and the binary restenosis rate was about half (4.7% for XIENCE vs. 8.9% for TAXUS at eight month follow-up. The rate for stent thrombosis at 284 days was 0.46%.
Boston Scientific has mapped out a timetable for introduction of new coronary stents extending to 2011 — with the very large assumption that it can satisfy the FDA’s concerns in a warning letter that has thus far blocked any new approvals. The new products encompass essentially all of the technological approaches being pursued in this segment. The company plans to introduce at least one new product each year from 2007 to 2011 in its paclitaxel-eluting stent family, starting with the Libert stent in 2007, followed by the Promus — the company’s branded version — through an agreement with Abbott, of the Xience.
In 2009, the Element stent will be launched, which will represent the first device based on the new Odyssey platform. The Odyssey platform employs a new alloy, platinum-enhanced stainless steel, as well as a stent geometry designed for optimum drug delivery. The stent will feature multiple drug elution, bioactive surface technology, and new polymers.
While many other suppliers are migrating to cobalt/chromium alloys for coronary stents, Boston Scientific has concluded that its new alloy, based on proven stainless steel but modified to optimize certain characteristics, will out-perform cobalt-chromium. The company plans launch of its Petal stent for use in bifurcation lesions in 2010 or 2011, and is targeting introduction of a bioabsorbable stent in 2011. Boston Scientific has already established a relationship with REVA Medical in the area of absorbable stents.
COURAGE offers large challenge
Mainstream media are attracted to bad news, and they got it from this year’s ACC meeting. National media outlets around the country, both newspapers and electronic, headlined the results of the COURAGE trial rolled out at the meeting and promptly sent the fairly un-nuanced message — to the general public at least — that stenting is no better than regimens of drugs and exercise. And it is possible that the stent designs will receive some heavy questioning concerning their ability to address the issues raised COURAGE, since lowering rates of late stent thrombosis will have a minor effect on overall clinical outcomes, such as cardiac death and myocardial infarction rates.
The results of COURAGE, a randomized trial of patients with stable coronary artery disease which involved 1,006 subjects who received a BMS device, along with optimal medical therapy and 1,210 treated with optimal medical therapy only, were presented at a press conference by William Boden, MD, of Buffalo General Hospital (Buffalo, New York).The data showed no significant difference in the rate of death, myocardial infarction, or stroke between the two groups. The 4.6-year cumulative primary rates of death or non-fatal heart attack were 19% and 18.5% in the percutaneous intervention and medical therapy groups respectively.
While DES were not used in the majority of PCI patients in COURAGE — since the devices were not approved in the U.S. until the last six months of the study — Boden argued that other randomized trials have already demonstrated that DES do not improve clinical outcomes such as death and myocardial infarction compared to BMS devices, indicating that the conclusions of the trial would be the same if DES had been used.
While the investigators stated that PCI has proven clinical benefit of improved survival in patients with MI compared to medical therapy — and also is better at least in the short term at relieving angina symptoms — they did not find a clinical benefit in patients with stable disease with respect to major clinical endpoints. Furthermore, a health status and economic outcomes sub-study of COURAGE patients found PCI combined with optimal medical therapy to be almost twice as expensive over three years ($11,960 for PCI vs. $6,665 for medical therapy), with equivalent quality of life.
Boden was unable to provide an estimate of the percentage of patients in a typical cardiology practice who would fall into the category of those treated with PCI in the COURAGE trial but indicated it would be a significant proportion of the total. However, Boden also said that he did not expect the results of the COURAGE trial to have a major impact on utilization of PCI.
Other experts commenting on the COURAGE results at the conference noted that previous trials such as RITA and MASS2 had produced similar findings but with no follow-on impact on practice. This may because PCI has a role primarily when medical therapy fails, as well as in high-risk patients with ischemic coronary artery disease where a benefit vs. medical therapy has been demonstrated.
Anti-platelet attention increases
The heightened focus on safety of DES elevated the level of interest in another topic that attracted considerable attention, both the problem of maintaining patient compliance for an expensive regiment of pharma therapy and also the variability of patient response to anti-platelet therapy.
Anti-platelet therapy, typically consisting of daily oral doses of aspirin and clopidogrel, has been a key factor in improving the safety of stent procedures. Treatment with aspirin alone as an anti-platelet agent, as is typically done for patients undergoing medical therapy, proved inadequate to prevent episodes of early stent thrombosis in the BMS era, since coverage of the stent with endothelial cells typically requires a few weeks to occur, at which point the risk of blood clot formation on the metal surface disappears.
With DES, however, endothelial cell coverage is delayed, resulting in the need to extend dual anti-platelet therapy, now recommended for one year unless there are contraindications. However, a significant percentage of patients — estimated as high as 25%-30% by some — do not respond normally to anti-platelet therapy due to aspirin or clopidogrel resistance.
As discussed by Steven Steinhubl, MD, of Gill Heart Institute at the University of Kentucky (Louisville) at the sessions, publications on aspirin resistance have increased exponentially over the past few years. Increasing dosages of anti-platelet therapy, particularly through the process known as re-loading — the re-administration of the initial 600 mg loading dose of clopidogrel — has been shown to improve response in resistant patients.
However, higher doses can also lead to bleeding problems, as demonstrated by data from the PCI-CURE study discussed by Sanjit Jolly, MD, of Hamilton Health Sciences (Hamilton, Ontario) at an ACC press conference. According to Jolly, there is a two-fold increase in bleeding risk for high-dose aspirin therapy, post-PCI, compared to low-dose aspirin, whereas rates of thrombosis do not appear to differ with dose based on comparison of patients in the U.S. vs. Europe. As a result, clinicians are searching for approaches that will allow them to detect resistance to aspirin and clopidogrel, so that high-dose therapy can be reserved for those patients who will actually benefit.
Anti-platelet assessment via devices
A number of devices are on the market or under development for assessing aspirin and clopidogrel resistance, including the VerifyNOW P2Y12 and Ultegra system from Accumetrics (San Diego), the Platelet Aggregation Profiler from BIO/DATA (Horsham, Pennsylvania), the Multiplate platelet function analyzer from Dynabyte Informationssysteme (Munich, Germany), the Plateletworks analyzer from Helena Labs (Beaumont, Texas), the Impact-R from DiaMed (Cressier sur Morat, Switzerland), and the PFA-100 from Dade Behring (Deerfield, Illinois).
The most recent ACC guidelines state that platelet function tests performed with such systems can be used to adjust clopidogrel dose in patients who at risk of a catastrophic event if sub-acute thrombosis were to occur.
Experts who discussed the use of platelet function testing at the conference, such as Steinbuhl, concluded that there is no firm evidence that altering anti-platelet therapy based on platelet function or aspirin resistance test results impacts clinical outcomes. However, data presented at the onference, such as results from the RECLOSE trial, showed a significant difference in event rate between responders and non-responders to clopidogrel (2.3% vs 8.6% stent thrombosis rates respectively) for patients classified as having probable stent thrombosis (although not for those classified as having definite stent thrombosis).
As discussed by Adnan Kastrati, MD, of Deutsches Herzzentrum (Munich, Germany), there is a high degree of correlation between late stent thrombosis and clopidogrel response, as well as for cardiac death and MI. The consensus is that tests for clopidogrel and aspirin resistance are likely to have value in selecting the optimal dose for individual patients but that data from large-scale randomized trials are still much needed to validate their use and to define algorithms for dose adjustment.
Revalving percutaneously
In contrast to the challenges facing stenting, there is no debate concerning the need to push aggressively forward with development of one of the next breakthrough areas of cardiovascular therapy: the need to repair or replace heart valves interventionally, thus avoiding the trauma of open surgery and expanding the therapy to high-risk groups of patients, primarily the elderly. And so the latest research focused on percutaneous therapy for heart valve disorders was another major topic at the scientific sessions.
Heart valve disorders have a high prevalence worldwide. Based on a study published by Francesca Bursi, MD et al. of the Mayo Clinic and Foundation (Rochester, Minnesota), 50% of myocardial infarction patients have mitral regurgitation (MR), with 12% of cases classified as moderate to severe. And two-thirds of those patients with moderate to severe MR will go on to develop heart failure. Percutaneous heart valves, or more generally transcatheter heart valves, could potentially drive a significant expansion of the market for heart valve therapy devices, now estimated by suppliers at $1.2 billion worldwide, because a significant proportion of patients with heart valve disorders (perhaps over half of patients in the U.S.) are not currently treated surgically.
Reasons for foregoing surgical treatment include patient refusal of a surgical procedure, lack of symptoms, and patients who are too ill to undergo surgery. In many of the first percutaneous implant procedures performed in France, only patients who were not surgical candidates were treated, since use of the percutaneous procedure reduces procedural risk and morbidity significantly.
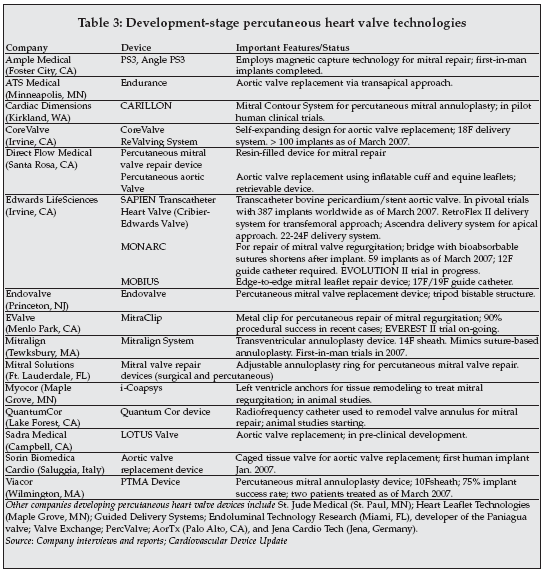
Numerous companies are developing devices for percutaneous repair and replacement of heart valves, mainly focusing on treatment of mitral and aortic valve disorders, as shown in Table 3. The total number of patients treated so far in clinical trials is approaching 400.
 |
John Webb, MD, of the University of British Columbia (Vancouver), whose institution has now treated over 100 patients with percutaneous aortic valve implants, has found that patients with the most severe dysfunction derive the greatest benefit from the procedure. Webb also noted, however, a significant learning curve for percutaneous implant procedures: the success rate for the first 25 procedures attempted was only 76%, but the rate increased to 96% for the second 25.
A very positive finding is that the death rate for patients treated with percutaneous techniques in Webb’s experience is 12% vs. a predicted death rate of 28% for patients managed with conventional therapy. Furthermore, the death rate dropped from 16% in the first 25 patients to 8% in the second 25. The statistics from Webb’s group are consistent with those from other centers implanting the Cribier-Edwards valve.
Percutaneous valve technologies are changing rapidly, including development of new delivery systems that simplify the technique and diminish the learning curve. In addition, developers are focusing on reducing the diameter of the delivery system, since most devices now require use of a guide catheter or introducer sheath ranging from 10 Fr-24 Fr, depending on design, type and size of the valve.
Edwards LifeSciences, the leader in the surgically implanted tissue heart valve market, is also leading in the percutaneous valve segment, with three different devices under developmentn (See more on Edwards’ valve focus, p., 13). CoreValve is another leader in the race to bring percutaneous valve technology to the market, with more than 100 implants completed with its ReValving System.
Transfemoral or transapical?
An emerging area of controversy in transcatheter valve development is the merits of transfemoral vs. transapical delivery. Kenton Zehr, MD, of the Heart, Lung, and Esophageal Surgery Institute, University of Pittsburgh Medical Center, discussed the topic in an ACC session on percutaneous heart valve therapy. The transapical approach, which typically is performed via a mini-thoracotomy, can be accomplished either by using beating heart or stopped-heart techniques, i.e., either with or without extracorporeal circulation. Consequently, procedures performed via a transapical approach fall into the realm of the cardiac surgeon, whereas those using a transfemoral approach are more suited to the interventional cardiologist.
Zehr argued that the transapical approach avoids risk of damage to vascular structures that must be crossed using the transfemoral approach, and also allows use of larger-diameter and stiffer delivery catheters which, at least with existing technologies, facilitates precise placement of the valve.
Edwards has responded by developing devices for both approaches, allowing physicians to determine the optimum delivery technique. As transcatheter valve technology continues to advance, addressing the issues with existing transfemoral devices, there is likely to be continued pressure, both from physicians and patients, for use of transfemoral techniques to make procedures truly minimally invasive. Developers, however, are likely to develop devices for both approaches whenever possible, expanding the range of physicians who can adopt the technology.
Development of transcatheter valve products is also likely to prove challenging from a regulatory perspective. Current FDA guidelines for heart valve device approval, for example, are written for surgically implanted valves, and do not address topics such as delivery systems, although some relevant guidance may be included in documents for coronary stents. Furthermore, recent issues related to long-term adverse events with devices such as coronary stents and ICDs will make regulators demand longer follow-up of patients receiving transcatheter valves.
The FDA is now recommending use of on-line clinical event reporting even in clinical trials, and long-term (5-10 year) monitoring to evaluate issues such as heart failure progression and collateral effects on remaining mitral or aortic valve structures in patients who receive transcatheter valves. Nevertheless, the field is progressing rapidly, and could emerge as a major new market in the cardiovascular device sector within the next one to two years.